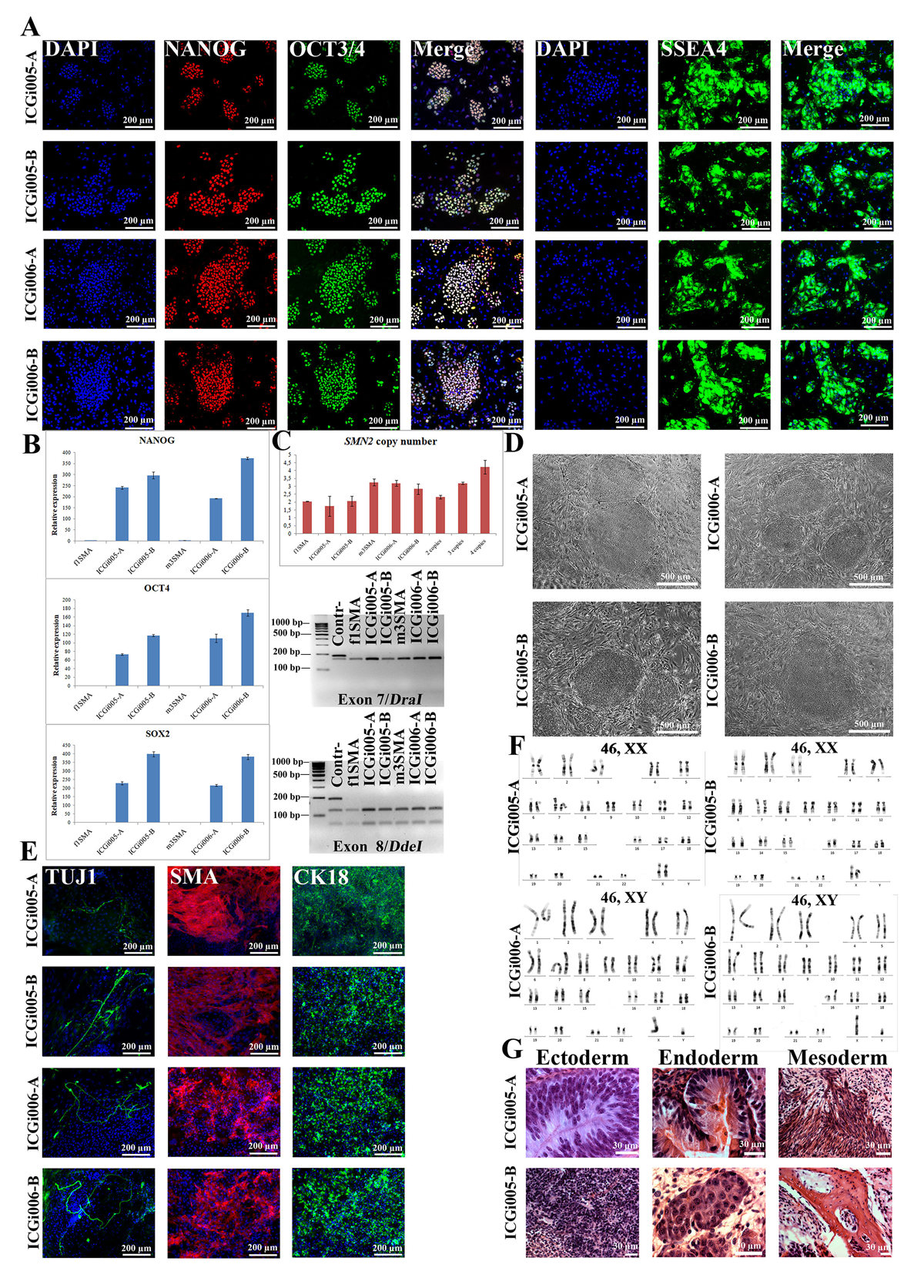
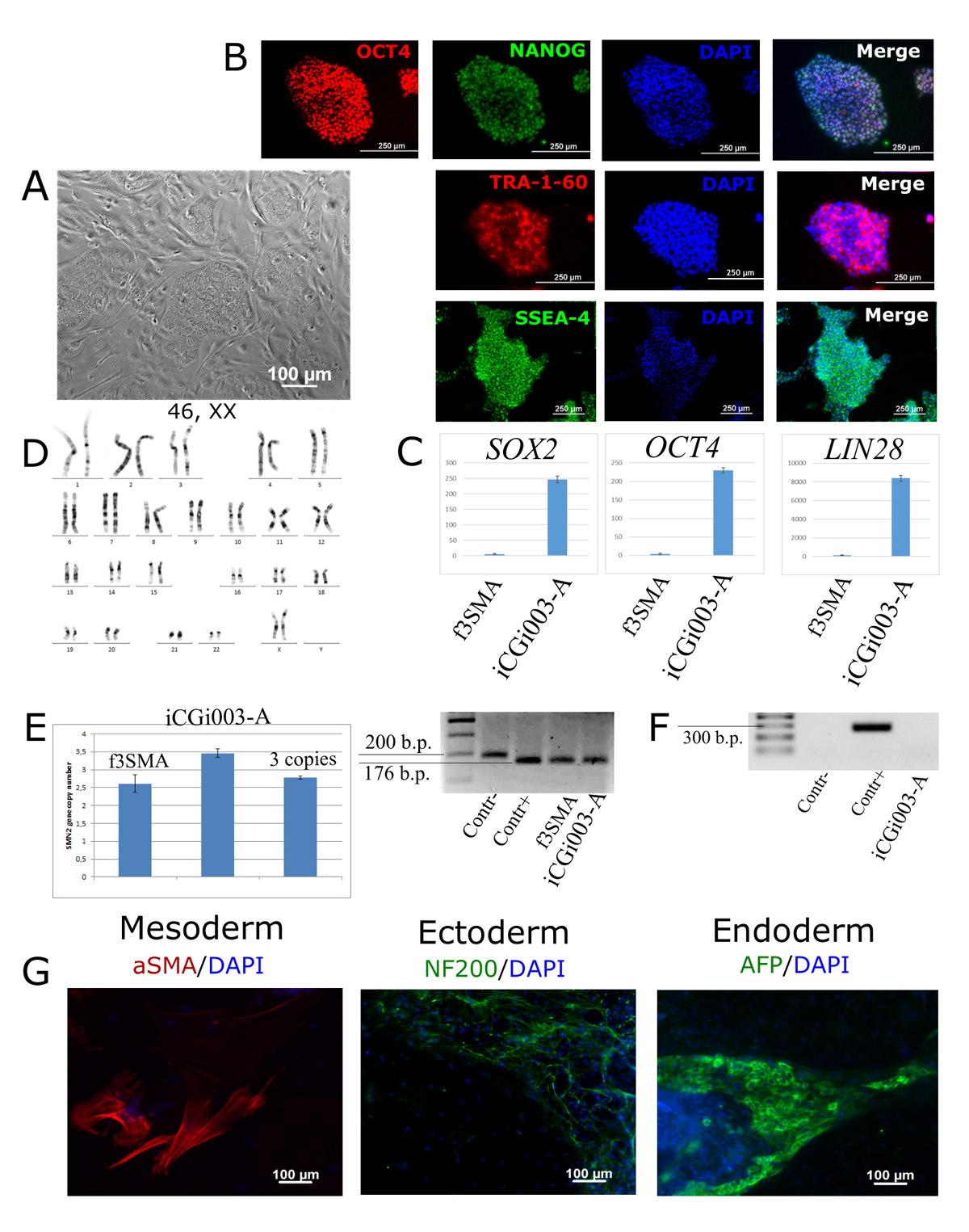
Spinal muscular atrophy (SMA) is characterized by the degeneration of motor neurons in the anterior horn of the spinal cord, which leads to striated muscle weakness. SMA is an autosomal recessive disease caused by deletion, mutation, or rearrangement of the SMN1 gene. However, the amplification of its nearly identical gene SMN2 promotes the alleviation of the disease due to the increasing level of SMN (survival motor neuron) protein. The severity of SMA depends on the SMN2 copy numbers:
- SMA type I is the most severe and spread type of the disease. Patients have one or two copies of the SMN2 gene, and their condition is characterized by inability to sit and control head muscles. The disease manifests before 6-month age, and the vast majority of the patients dies before 2-years age due to the respirator muscles atrophy.
- SMA type II patients usually have 3 copies of the SMN2 gene. This type of the disease manifests between 6- and 18-month age, and the patients are able to sit and stand by themselves, but they cannot walk.
- SMA type III includes variative manifestations, but the patients are able to walk. The duration of their lives has no significant decrease. There are 3 or 4 copies of the SMN2 gene in the patients’ genomes.
Although the pathogenesis bases are well observed, the molecular pathways of the disease remain unclear. The investigation of pathological processes in motor neurons is difficult because of the inability to safely extract them from patients as well as the postmortal neurons present the information only about the terminal stage of the disease. To cover these problems, patient’s cells can be reprogrammed to a pluripotent state with further differentiation into the target motor neurons. Thus, we generated SMA type I (https://hpscreg.eu/cell-line/ICGi005-A), II (https://hpscreg.eu/cell-line/ICGi006-B) and III (https://hpscreg.eu/cell-line/ICGi003-A) induced pluripotent stem cell. The iPSC lines can be used for further studies by providing in vitro the relevant cell types.