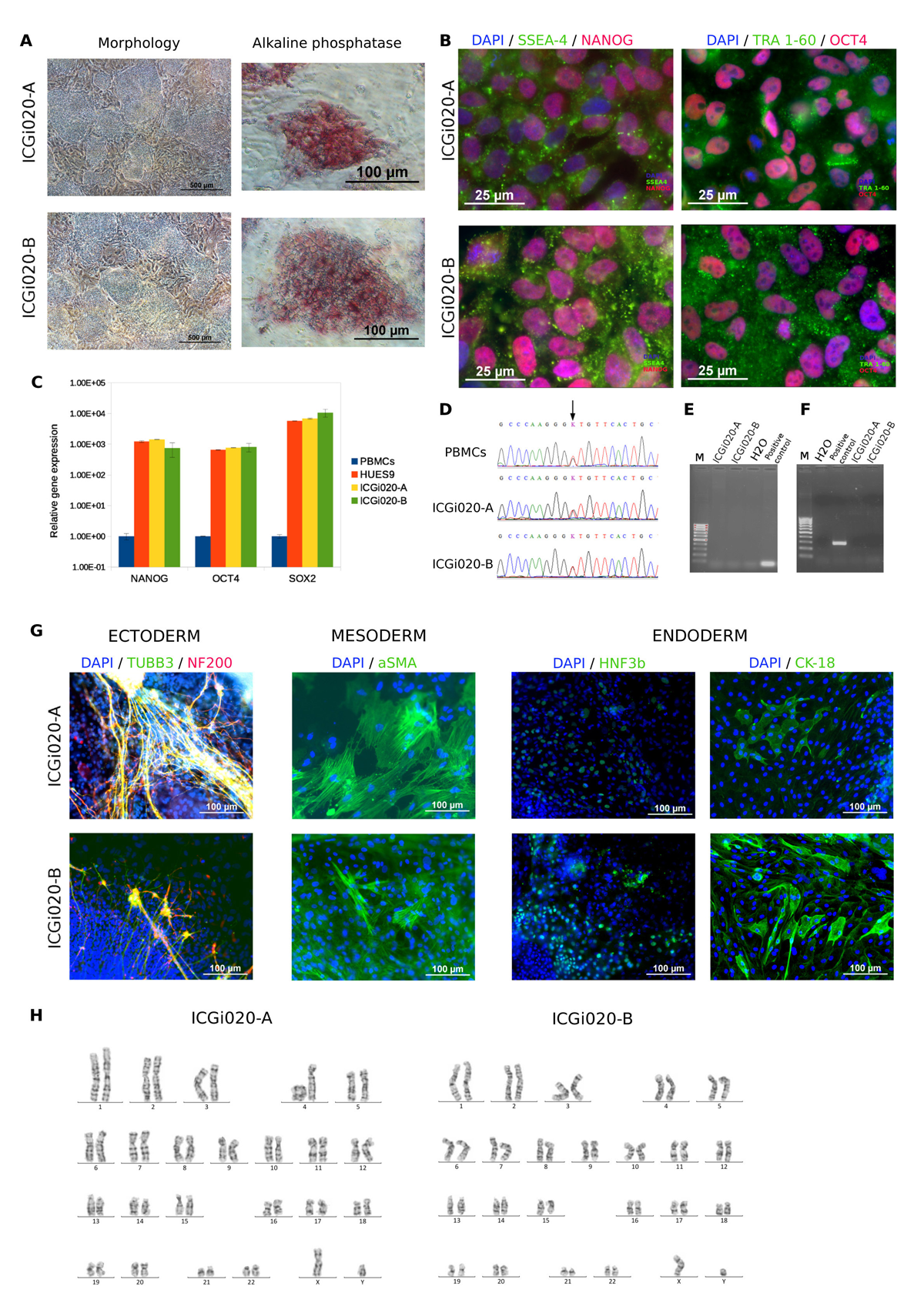
Wilson’s disease is an inherited disorder associated with copper accumulation in the liver, brain and other vital organs. Wilson’s disease is caused by mutations in the ATP7B gene. Over 300 mutations of ATP7B have been described. Despite the disease is autosomal recessive, the patient whose PBMCs were reprogrammed in the study harbours heterozygous mutation c.3207C>A (p.H1069Q). Detailed analysis of the ATP7B complete gene sequencing data has not revealed other known disease associated mutation. The generated iPSC lines maintained the original genotype, expressed pluripotency markers, had normal karyotype and demonstrated the ability to differentiate into derivatives of the three germ layers. The patient-specific iPSC lines are useful for modelling the disease in vitro and searching for modifier genes that cause the phenotypic heterogeneity. Studying pathogenetic copper metabolism pathways in hepatocyte-like cells derived from the iPSCs is helpful for discovering targeted therapy of the chronic progressive disease.